Abstract
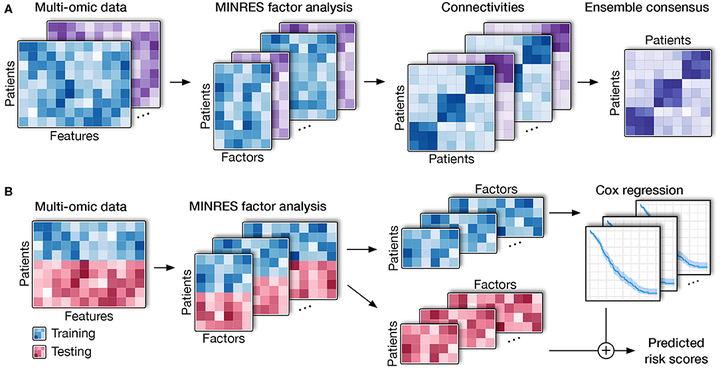
Cancer is an umbrella term that includes a range of disorders, from those that are fast-growing and lethal to indolent lesions with low or delayed potential for progression to death. One critical unmet challenge is that molecular disease subtypes characterized by relevant clinical differences, such as survival, are difficult to differentiate. With the advancement of multi-omics technologies, subtyping methods have shifted toward data integration in order to differentiate among subtypes from a holistic perspective that takes into consideration phenomena at multiple levels. However, these integrative methods are still limited by their statistical assumption and their sensitivity to noise. In addition, they are unable to predict the risk scores of patients using multi-omics data. Here, we present a novel approach named Subtyping via Consensus Factor Analysis (SCFA) that can efficiently remove noisy signals from consistent molecular patterns in order to reliably identify cancer subtypes and accurately predict risk scores of patients. In an extensive analysis of 7,973 samples related to 30 cancers that are available at The Cancer Genome Atlas (TCGA), we demonstrate that SCFA outperforms state-of-the-art approaches in discovering novel subtypes with significantly different survival profiles. We also demonstrate that SCFA is able to predict risk scores that are highly correlated with true patient survival and vital status. More importantly, the accuracy of subtype discovery and risk prediction improves when more data types are integrated into the analysis. The SCFA software and TCGA data packages will be available on Bioconductor.